

A atrofia muscular espinhal (AME) é uma doença genética progressiva que afeta aproximadamente um em cada 10 mil nascidos vivos no mundo, sendo a principal causa genética de óbitos em bebês.
Em indivíduos com AME, a falta de uma proteína importante, a SMN, leva à morte de neurônios que transmitem impulsos para os músculos. Os sinais cerebrais não chegam à musculatura, que se enfraquece e atrofia, resultando em problemas graves que afetam funções básicas do organismo, como respirar, andar, falar e se alimentar.
Os sintomas da AME geralmente se manifestam na infância e vão se agravando rapidamente com o tempo. Caso não haja tratamento, a maioria das crianças não atinge os dois anos de idade. Por isso, o diagnóstico e, principalmente, tratamento precoces, são essenciais para garantir uma melhor qualidade de vida para a criança.
Felizmente, as pesquisas científicas, intensificadas pelo estudo do genoma e as novas tecnologias de manipulação do DNA, permitiram o desenvolvimento e avanço de terapias que impactam o histórico natural da doença e melhoram a qualidade de vida das pessoas com AME.
Conheça os marcos históricos dessa doença a seguir.
Atrofia muscular espinhal – 130 anos de pesquisas científicas
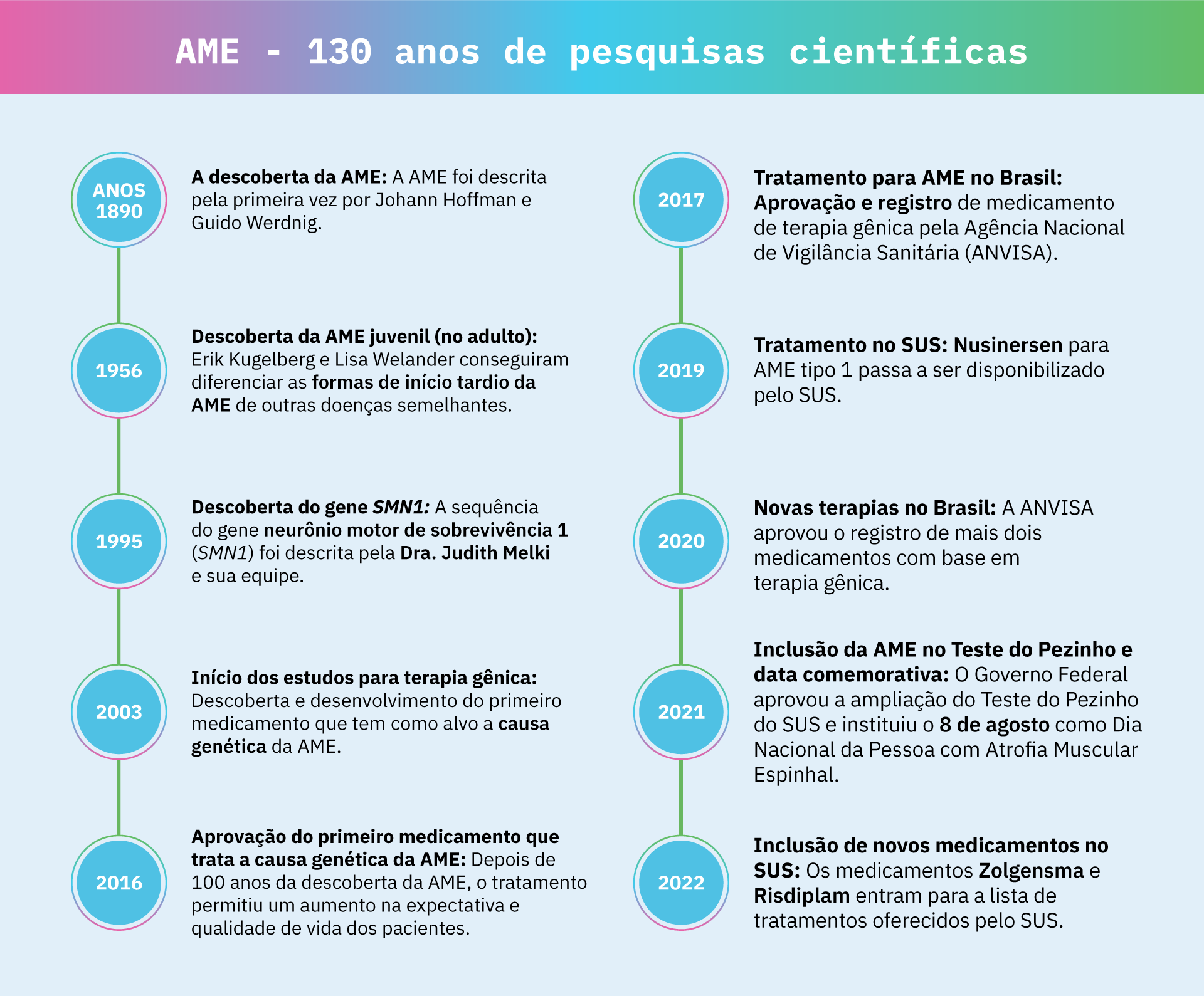
Anos 1890s – A descoberta da AME
A AME foi descrita pela primeira vez por dois cientistas, Johann Hoffman e Guido Werdnig, que observaram vários casos de bebês que desenvolveram fraqueza muscular nos primeiros meses de vida. Eles perceberam que as células do neurônio motor desses bebês pareciam degenerar e também notaram que essa condição parecia ser familiar (hereditária).
Os estudos desses dois cientistas levaram à identificação da AME e por isso, até hoje, a AME infantil (tipos 0 e 1) é conhecida como doença de Werdnig-Hoffman.
1956 – Descoberta da AME juvenil (no adulto)
Algumas décadas depois, dois outros cientistas, Erik Kugelberg e Lisa Welander, conseguiram diferenciar as formas de início tardio da AME de outras doenças semelhantes, como a distrofia muscular. Por isso, a AME tipo 3 também é chamada doença de Kugelberg-Welander.
1995 – Descoberta do gene SMN1
A sequência do gene neurônio motor de sobrevivência 1 (SMN1) foi descrita pela Dra. Judith Melki e sua equipe, que também mostraram que alterações nesse gene estavam presentes em pessoas com AME. Além disso, a equipe identificou o gene SMN2, associado às diferentes gravidades da AME.
Acontecimentos importantes
A descoberta do gene SMN1 foi fundamental para permitir o diagnóstico genético da AME. Antes disso, o diagnóstico era feito com base nos sinais e sintomas clínicos do paciente.
A descoberta do SMN1 e SMN2 permitiu o estudo e desenvolvimento de tratamentos para AME.
2003 – Início dos estudos para tratamentos com terapia gênica
Quatro anos após a identificação do SMN2, outro grupo de pesquisadores descreveu as alterações nesse gene que estão associadas à AME. A partir dessas descobertas, estudos visando a terapia gênica para a doença se iniciaram.
Esses estudos levaram à descoberta e ao desenvolvimento do primeiro medicamento da AME, o Nusinersen (nome comercial: Spinraza), que tem como alvo a causa genética da doença, ajudando o organismo a produzir mais proteínas SMN a partir do SMN2.
2016 – Aprovação do primeiro medicamento que trata a causa genética da AME
A comunidade global da AME comemorou a aprovação do primeiro tratamento baseado em terapia gênica, o Nusinersen, pela Food & Drug Administration (FDA) nos Estados Unidos.
| Depois de 100 anos da descoberta da doença, este foi um marco histórico, possibilitado pelo empenho dos pesquisadores, investimentos e apoio da sociedade. Até então, a expectativa de vida de pessoas com AME era muito baixa, mas com o novo tratamento há melhora significativa na qualidade de vida. |
2017 – Aprovação de tratamento para AME no Brasil
Pacientes brasileiros puderam comemorar a aprovação e registro do Nusinersen pela Agência Nacional de Vigilância Sanitária (ANVISA).
2019 – Tratamento no SUS
O Ministério da Saúde inseriu o Nusinersen para AME tipo 1 no Protocolo Clínico e Diretrizes Terapêuticas (PCDT), disponibilizando o tratamento pelo SUS.
Crianças com os tipos 2 e 3 podem obter o tratamento na modalidade compartilhamento de risco, onde o governo paga pelo medicamento somente se houver melhora da saúde do paciente.
2020 – Novas terapias aprovadas no Brasil
A ANVISA aprovou o registro de dois outros medicamentos com base em terapia gênica, que corrigem a falta do SMN1 e do SMN2, o Onasemnogene abeparvovec (nome comercial: Zolgensma) e o Risdiplam (nome comercial: Evrysdi).
2021 – Inclusão da AME no Teste do Pezinho e data comemorativa
O Governo Federal aprovou o Projeto de Lei (PL 5.043/2020) de ampliação do Teste do Pezinho do SUS. De seis doenças, a triagem contará com 53 doenças, incluindo AME.
A triagem neonatal da AME representa um enorme avanço para o tratamento da doença, que é progressiva e, por isso, necessita de um diagnóstico precoce. Quanto antes for realizado o diagnóstico, mais cedo o tratamento pode ser iniciado, evitando o avanço e agravamento dos sintomas.
Além disso, o Governo Federal também sancionou a Lei 14.062, que institui o 8 de agosto como Dia Nacional da Pessoa com Atrofia Muscular Espinhal.
2022 – Inclusão de novos tratamentos de atrofia muscular espinhal pelo SUS
Em 2022, o Ministério da Saúde anunciou a inclusão do Zolgensma e Risdiplam na lista de medicamentos disponíveis para o tratamento de AME oferecidos pelo SUS no Brasil.
2025 – Aplicação de um dos medicamentos mais caros do mundo pelo SUS
Em 2025, o SUS viabilizou a aplicação do medicamento Zolgensma, um dos mais caros do mundo, para pacientes com AME tipo 1, seguindo critérios específicos.
Todo tratamento deve ser acompanhado por um médico e precisa ser adequado às necessidades de cada paciente.
O Teste da Bochechinha e a atrofia muscular espinhal
O Teste da Bochechinha é um teste de triagem neonatal genética que identifica mais de 530 doenças genéticas graves, incluindo AME, e que complementa o Teste do Pezinho oferecido pelo Sistema Único de Saúde – SUS.
No Teste da Bochechinha, o DNA do recém-nascido é analisado por Sequenciamento de Nova Geração (NGS) e são identificadas variantes que aumentam o risco para o desenvolvimento das doenças avaliadas.
Por ser um teste de triagem, o Teste da Bochechinha é indicado para recém-nascidos assintomáticos. Quando uma pessoa, de qualquer idade, tem sintomas de AME, o médico deve recomendar um exame genético de diagnóstico para confirmar a suspeita clínica.
A Mendelics, laboratório que criou o Teste da Bochechinha, oferece diferentes exames genéticos para diagnóstico da AME, incluindo o MLPA dos genes SMN1 e SMN2, o Painel de Doenças Tratáveis e o Exame de Sequenciamento do gene SMN1.
O Teste da Bochechinha não precisa de pedido médico para ser realizado, já exames de diagnóstico genético necessitam do pedido de um especialista.
Tem alguma dúvida sobre a AME ou sobre o Teste da Bochechinha? Deixe sua pergunta nos comentários abaixo ou entre em contato com a nossa equipe pelo telefone (11) 5096-6001 ou através do nosso site.
É médico e quer oferecer o Teste da Bochechinha para seus pacientes? Entre em contato e seja nosso parceiro.
Texto escrito por: Ágatha Faria
Revisado e atualizado por: Nathália Taniguti, em julho de 2025
Referências
- Prior TW, Leach ME, Finanger E. Spinal Muscular Atrophy. 2000 Feb 24 [Updated 2019 Nov 14]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1352
- Talbot K, Tizzano EF. The clinical landscape for SMA in a new therapeutic era. Gene Ther. 2017;24(9):529-533.
- https://www.curesma.org/the-discovery-of-sma/
- https://portalarquivos2.saude.gov.br/images/pdf/2019/outubro/23/Portaria-Conjunta-PCDT-Atrofia-Muscular-Espinhal-5q-Tipo-I-final.pdf
- https://saude.gov.br/saude-de-a-z/atrofia-muscular-espinhal-ame
- https://iname.org.br/tratamentos-da-ame/visao-geral/
Revisão
A atrofia muscular espinhal (AME) é uma doença neuromuscular, de origem genética, que afeta os neurônios motores da medula espinhal e do tronco cerebral. A morte dos neurônios motores provoca fraqueza e atrofia muscular e pode afetar funções básicas do organismo.
Alterações no gene SMN1 causam a atrofia muscular espinhal. O gene SMN1 produz a proteína SMN, importante para a manutenção dos neurônios motores.
Sim, recentemente o Ministério da Saúde anunciou a inclusão de dois novos medicamentos para tratamento de AME, a serem disponibilizados pelo SUS, o Onasemnogene abeparvovec (nome comercial: Zolgensma) e o Risdiplam (nome comercial: Evrysdi).
0 comentários