

As mucopolissacaridoses (MPS) são um grupo de doenças raras de origem genética que fazem parte das doenças lisossômicas.
Elas fazem parte dos erros inatos do metabolismo, um grupo de doenças genéticas, hereditárias, que causam o mau funcionamento de alguma via metabólica, resultando em níveis alterados de algumas substâncias importantes para o bom funcionamento do organismo.
Apesar dos sintomas das mucopolissacaridoses estarem presentes desde o nascimento, o diagnóstico demora, em média, quatro a cinco anos para ser atingido. Por ser uma doença rara e pouco conhecida, o diagnóstico costuma ser desafiador, uma vez que os primeiros sinais são muito inespecíficos e podem ser confundidos com outras doenças.
| Dia 15 de maio é celebrado o Dia Internacional da Conscientização da MPS. Conhecido como #MPSDay, a data tem o objetivo de conscientizar a população, profissionais da saúde e os médicos sobre os cuidados relacionados às mucopolissacaridoses e a importância do diagnóstico e tratamento precoces. |
Quais os sinais e sintomas da MPS?
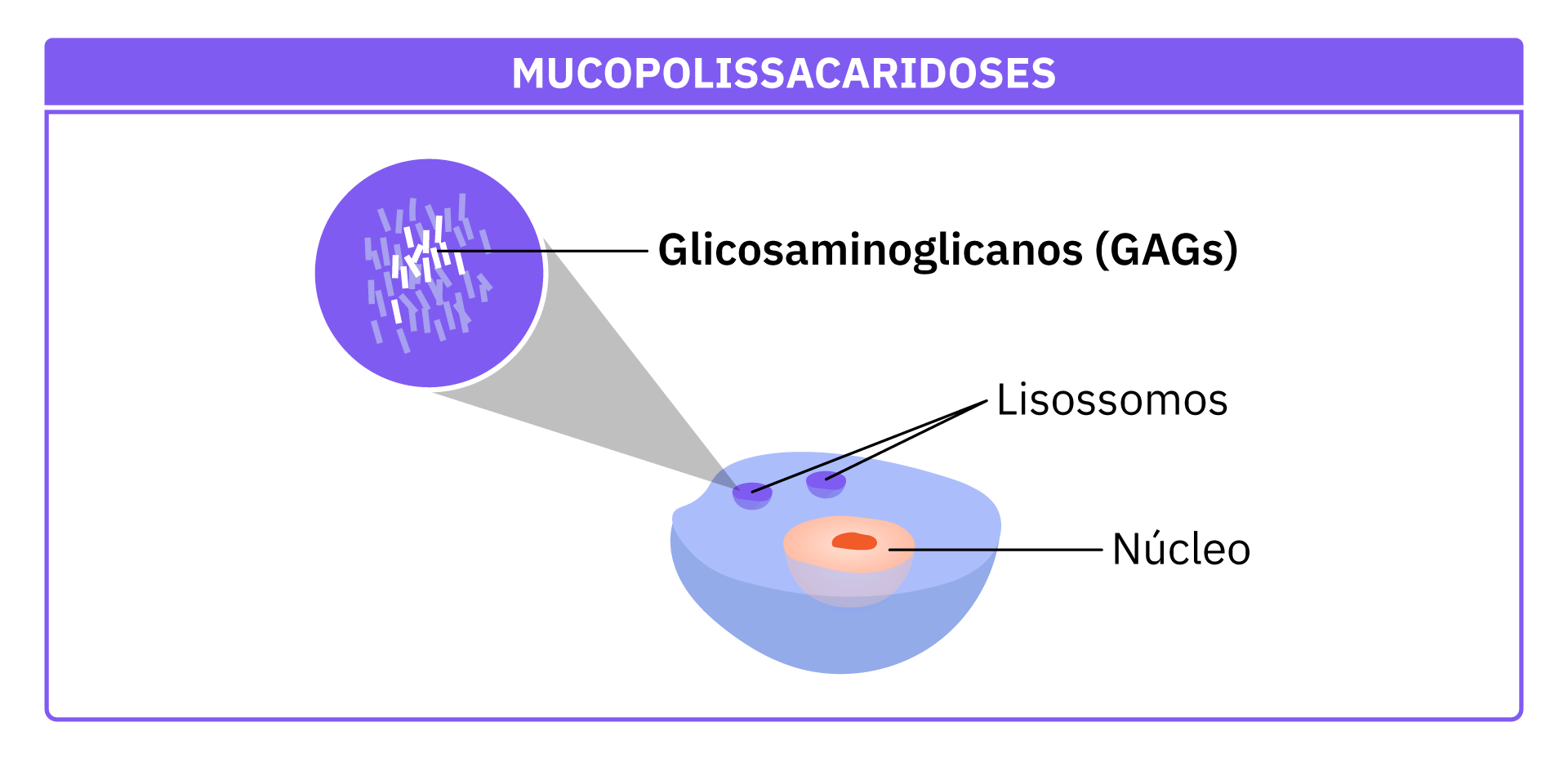
Nas MPS, há uma formação inadequada de enzimas responsáveis por quebrar açúcares complexos chamadas glicosaminoglicanos (GAGs), encontradas nos lisossomos (compartimento celular responsável por digerir e reciclar moléculas). Com isso, os GAGs se acumulam nos lisossomos das células, provocando sintomas em todo o organismo, principalmente nas artérias, esqueleto, olhos, articulações, orelhas, pele e/ou dentes.
Existem vários tipos e subtipos diferentes de MPS:
- MPS I – Síndrome de Hurler, Síndrome de Hurler-Scheie ou Síndrome de Scheie
- MPS II – Síndrome de Hunter
- MPS III – Síndrome de Sanfilippo
- MPS IV – Síndrome de Mórquio
- MPS VI – Síndrome de Maroteaux-Lamy
- MPS VII – Síndrome de Sly
- MPS IX – Deficiência de Hialuronidase
Os sinais, sintomas e a gravidade dos diferentes tipos de MPS pode variar muito entre as pessoas com a doença, mesmo entre aquelas com o mesmo tipo de MPS, e até mesmo entre membros da mesma família.
Na forma mais grave da MPS I, conhecida como Síndrome de Hurler, os sintomas começam a aparecer nos primeiros meses de vida do paciente. Mas, na forma branda (Síndrome de Scheie), o diagnóstico geralmente é feito apenas a partir dos 4 anos.
Em geral, a MPS é silenciosa e os sintomas surgem por volta de um ou dois anos de idade. Os sinais e sintomas das MPS se manifestam em múltiplos órgãos e podem acometer principalmente os ossos e as articulações, o coração, o sistema respiratório, fígado, baço, e também causar problemas neurológicos e cognitivos.
Se não tratada, a doença progride gravemente, acometendo todo o funcionamento do organismo e as funções cognitivas da pessoa, impactando fortemente a qualidade de vida.
Como é feito o tratamento da MPS?
As MPS não tem cura, mas tem tratamento!
Atualmente, existem opções de tratamentos específicos através da terapia de reposição enzimática (TRE) , que repõe a enzima em falta no organismo do paciente.
No Brasil, o Sistema Único de Saúde (SUS) disponibiliza medicamentos para o tratamento das MPS tipo I (laronisade), II (idursulfase alfa recombinante), IV (alfaelosulfase), VI (galsulfase) e VII (alfavestronidase). Até o momento não há tratamento específico disponível para a Síndrome de Sanfillipo.
Além do tratamento medicamentoso, pacientes com MPS necessitam de acompanhamento por vários especialistas e equipe multidisciplinar, com realização periódica de exames laboratoriais, exames de imagem e avaliações clínicas. Também pode ser necessário fisioterapia, fonoaudiologia e outras terapias a depender dos sinais e sintomas de cada paciente.
Qual a causa da mucopolissacaridose?
As MPS são causadas por alterações em genes que produzem enzimas responsáveis por degradar os glicosaminoglicanos (GAGs).
Em pacientes com MPS, a falta ou redução das enzimas que degradam os GAGs faz com que eles se acumulem. ocasionando os sintomas sistêmicos. Os principais ocorrem nas artérias, esqueleto, olhos, articulações, orelhas, pele e/ou dentes; porém, com o tempo (e sem tratamento), podem ser vistos em praticamente todos os órgãos.
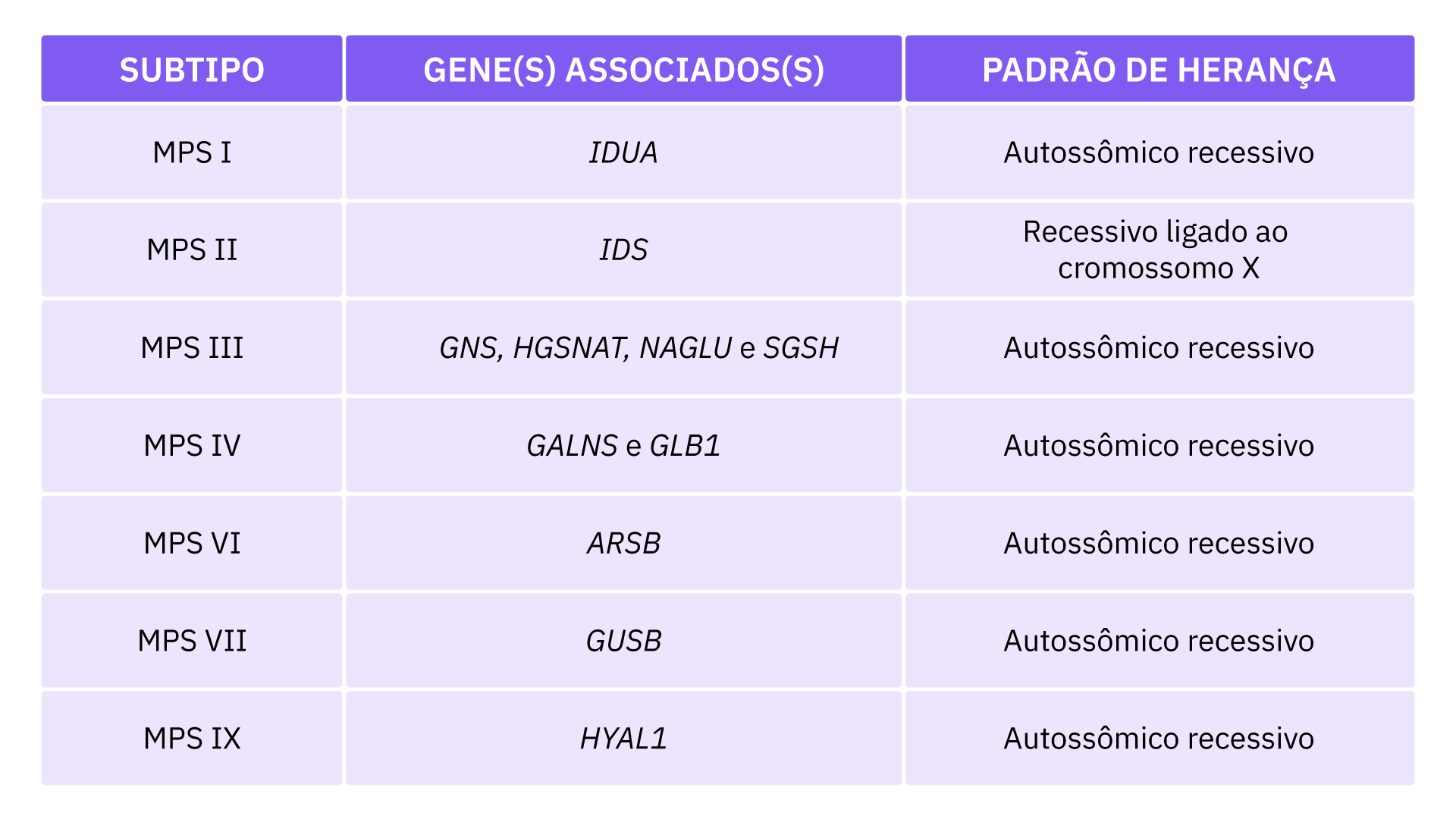
Com exceção da MPS tipo II, que possui padrão de herança ligado ao X recessivo, todas os outros subtipos do MPS são herdados de forma autossômica recessiva, isso quer dizer que o bebê nasce com a doença quando herda duas cópias alteradas de um gene, uma do pai e outra da mãe (mutação em homozigose).
Quando apenas uma cópia do gene alterado é herdada, a pessoa é chamada de portadora. Ela não vai ter a doença, mas pode transmitir a alteração para os filhos.
Entenda mais sobre padrões de herança e como as doenças genéticas são herdadas.
Na Síndrome de Hunter (MPS II), o gene alterado está localizado no cromossomo X, que é um dos dois cromossomos sexuais. Nos homens (que têm apenas um cromossomo X), uma cópia alterada do gene em cada célula é suficiente para causar a doença. Nas mulheres (que têm dois cromossomos X), uma mutação teria que ocorrer em ambas as cópias do gene para causar a MPS II.
É importante que seja feito aconselhamento genético em famílias com histórico de MPS para que os pais compreendam suas chances de ter outro filho com a doença.
Como é feito o diagnóstico de MPS?
Por ser uma doença rara e, consequentemente, pouco conhecida, o diagnóstico precoce costuma ser desafiador. Os primeiros sinais são muito inespecíficos e podem ser confundidos com outras doenças.
Quanto mais tarde o diagnóstico é feito, mais tarde o tratamento é iniciado, impactando diretamente a qualidade e expectativa de vida do paciente.
A suspeita de MPS é feita com base no exame clínico, e nos sintomas do paciente.
Podem ser realizados exames laboratoriais para detectar níveis anormais de GAGs e de enzimas lisossomais nas células.
A confirmação do diagnóstico da doença é feita pelo exame genético capaz de detectar alterações em um gene associado à doença. Nesse caso, os Painéis de Sequenciamento de Nova Geração (NGS) para doenças metabólicas e doenças genéticas de início precoce são indicados.
A realização do teste genético também é altamente recomendada para pessoas que tenham histórico familiar da doença.
Teste de Bochechinha e identificação precoce de MPS
O Teste de Bochechinha é um teste de triagem neonatal genética que analisa o DNA do bebê em busca de mutações associadas a mais de 340 doenças genéticas tratáveis da primeira infância, incluindo as mucopolissacaridoses. O teste pode ser realizado desde o primeiro dia de vida do bebê até ele completar um ano de idade. A coleta do DNA, através da saliva, pode ser feita em casa, pelos próprios pais, com segurança e rapidez.
A MPS não é avaliada no Teste do Pezinho Básico do SUS e na maioria dos testes de triagem neonatal ampliados na rede particular.
Converse com um médico de sua confiança e, se houver a necessidade de um exame genético, entre em contato conosco.
| Caso a criança já apresente sintomas e os pais desejem fazer um exame de diagnóstico genético, a Mendelics, laboratório que criou o Teste da Bochechinha, oferece exames para o diagnóstico de MPS, incluindo o Painel de Doenças Tratáveis e o Painel de Síndromes Clinicamente Reconhecíveis. |
Quer saber mais sobre a MPS e o Teste da Bochechinha? Deixe sua pergunta nos comentários abaixo ou entre em contato com a nossa equipe pelo telefone (11) 5096-6001 ou através do nosso site.
Escrito por: Ágatha Cristina
Revisado por: Cleandra Gregório, em maio de 2024.
Revisão
As mucopolissacaridoses (MPS) são um grupo de doenças metabólicas raras e graves causadas pela falta de enzimas que quebram substâncias chamadas glicosaminoglicanos (GAGs). Elas se manifestam com sintomas em várias partes do corpo, como ossos, articulações, olhos e pele.
Os sinais e sintomas das MPS podem variar amplamente, mas geralmente incluem problemas como deformidades ósseas, dificuldades respiratórias, problemas cardíacos, problemas de visão e audição, além de atrasos no desenvolvimento. Os sintomas podem afetar várias partes do corpo, como articulações, pele, olhos e sistema nervoso.
As MPS são causadas por mutações genéticas que levam à deficiência de enzimas responsáveis pela quebra de glicosaminoglicanos (GAGs). Esses GAGs se acumulam dentro das células devido à deficiência enzimática, causando danos aos tecidos e órgãos.
O diagnóstico das MPS geralmente envolve um exame clínico, exames laboratoriais para detectar níveis anormais de GAGs e de enzimas lisossomais, além de exames de imagem. A confirmação é feita por meio de testes genéticos que detectam mutações nos genes associados às MPS. O Teste da Bochechinha pode ser uma opção para a identificação precoce em bebês.
Referências
- Mucopolysaccharidoses. Rare Diseases.
- Mucopolysaccharidosis. Genetic and Rare Diseases.
0 comentários